

第一作者:Zhesheng Hua
通讯作者:Xiang Gao (高翔) Hao Song(宋浩)
通讯单位:浙江大学
成果简介
钒氧化还原液流电池(VRFB)在固定式高容量储能领域成为领跑者,在安全性、循环寿命和系统剩余价值(资源回收)方面显示出比锂离子电池更显著的优势。尽管如此,电解液的高制造成本仍然是VRFB扩大商业化的一大障碍。利用甲酸作为还原剂催化钒电解液的生产是一条有前景且经济可行的途径。然而,阻碍大规模电解液生产的主要瓶颈在于催化剂的活性和耐久性不足。浙江大学高翔院士&宋浩教授团队开发了一种核壳催化剂用于通过催化还原生产V3.5+电解质,其特征是Pd核被Pt壳包裹。值得注意的是,与商业Pt/C相比,Pd/Pt摩尔比为1:2的核壳催化剂的活性显著提高了2.4倍。Pd的掺入改变了甲酸在Pt/C上的氧化途径,相对削弱了与CO中间体的结合,从而增强了催化剂的抗中毒性。同时,Pt壳有效地缓解了Pd在甲酸氧化过程中的失活问题。Pd/Pt摩尔比为1:2的核壳催化剂在反应中没有金属浸出,在重复生产中保持了优异稳定的效率,被证明是生产高质量V3.5+电解液的高效催化剂。
相关成果以“A promising catalyst for efficient and stable production of high-performance V3.5+ electrolyte in vanadium redox flow batteries”为题发表在Journal of Energy Storage上。
研究背景
Pt和Pd两种贵金属在甲酸氧化(FAO)催化剂的研究中显示出优异的活性。与Pt基催化剂相比,Pd基催化剂对FAO的反应活性更高,但它们的稳定性有待进一步提高。在Pt和Pd中引入另一种金属以获得具有特定结构和组成的二元催化剂,可以通过双金属之间的电子、空间和几何效应来增强其催化性能。尽管这些催化剂在CO耐受性方面具有优势,但它们不能直接用于生产V3.5+电解液,这是因为钒电解质的高酸性条件导致金属浸出和失稳。Qian等人发现,与具有过渡金属核(Fe、Co和Ni)的核壳纳米颗粒相比,具有贵金属核(Pd、Ir和Os)的M@Pt核壳纳米颗粒在酸性介质中具有更高的溶解电位和优异的电化学稳定性。研究表明由类似电化学系列元素组成的核壳纳米粒子具有增强的耐久性。Pd和Pt具有相似的面心立方(fcc)结构和几乎相同的晶格常数,允许Pt原子在Pd上外延生长。
在这项工作中,浙江大学高翔院士&宋浩教授团队提出了一种利用Pd(核)-Pt(壳)催化剂(表示为Pd@Pt/C)批量连续生产V3.5+电解质的方法,与之前发表的文献相比,本研究表明催化剂的贵金属负载量显著降低。同时,在反应过程中每摩尔VO2+的贵金属使用量降低。考虑到目前铂的价格比钯高5-10%,开发的催化剂在电解液生产中具有竞争力。与商业Pt/C催化剂相比,Pd1@Pt2/C催化剂(Pd/Pt摩尔比1/2)显示出优异的催化活性。Pd1@Pt2/C与Pd/C和Pt/C催化剂相比,Pd1@Pt2/C催化剂对CO中毒的抗性更强。在重复生产中,Pd1@Pt2/与Pt/C相比,Pd1@Pt2/C表现出稳定的效率。通过催化制备的V3.5+电解液没有金属浸出,性能与通过电化学方法制备的电解液相当。
核心内容
1.V3.5+电解液催化生产性能的评价
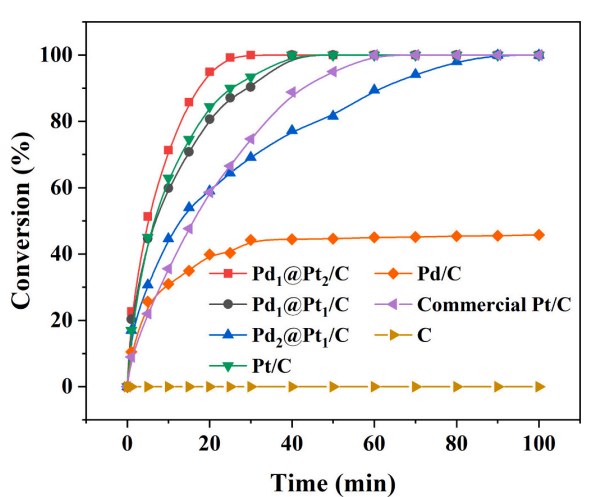
图1 在70℃下,不同催化剂在不同反应时间催化V4+转化为V3.5+(完全转化率对应V3.5+)
如图1所示,为比较不同摩尔比的Pdx@Pty/C催化剂的活性,Pt/C、Pd/C、市售的Pt/C和炭黑进行比较。据观察Pd1@Pt2/C表现出最高的催化活性,在反应25分钟后达到近100%的催化活性。Pd1@Pt1/C与Pt/C相当,而Pd2@Pt1/C表现出较差的活性。值得注意的是,即使在100分钟后,Pd/C催化剂的催化活性仍保持在50%以下,可能是由于Pd在甲酸氧化过程中的不稳定性和高负载Pd结块的趋势。当转化活性相对于金属质量和反应时间归一化时,Pd1@Pt2/C催化剂的峰值性能为12 mol⋅g−1⋅h−1,是商用Pt/C催化剂(5 mol⋅g−1⋅h-1)的2.4倍。
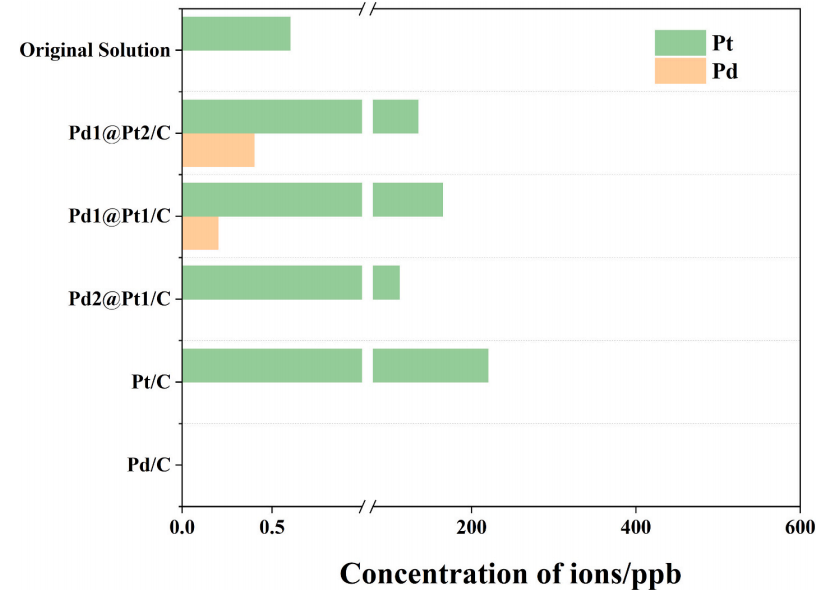
图2 在70℃下与各种催化剂进行60分钟催化反应后,通过ICP-MS定量溶解在V3.5+电解液中的金属离子浓度
催化剂的稳定性是催化剂设计中的关键参数。电解液中金属杂质离子的存在会在VRFB充电过程中引发负极上的析氢,从而对其性能产生不利影响。因此,利用电感耦合等离子体质谱法(ICP-MS)分析了催化后电解质中浸出的Pt和Pd离子的浓度,从而深入研究了Pdx@Pty/C、Pt/C、Pd/C的化学稳定性,图2总结了ICP分析的结果。对于每种催化剂,电解液中Pd离子的浓度保持在噪声水平内,而Pt离子的浓度保持为十亿分之250(ppb)以内。值得注意的是,从Pt离子溶解最多的Pt/C中浸出的Pt的质量损失率仅为0.002%。因此,在实际考虑中,反应过程中催化剂的浸出几乎可以忽略不计。
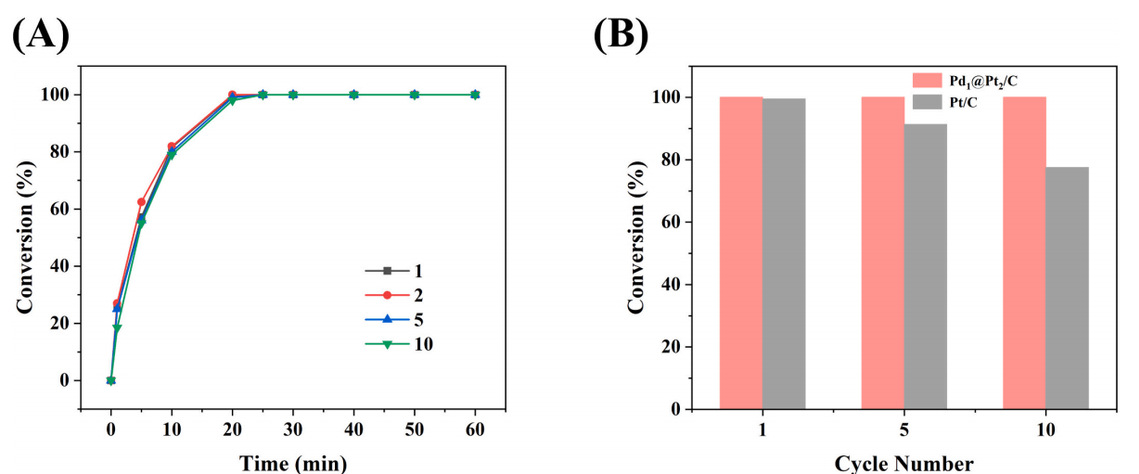
图3 (A)70℃下重复催化还原过程中转化率的时间演变(Pd1@Pt2/C不变);(B)Pd1@Pt2/C和Pt/C在70℃的重复实验中转换率(每次还原持续40分钟)
为评估催化剂在反应过程中的稳定性,对Pd1@Pt2/C和Pt/C催化剂的催化还原进行了重复实验用于监测它们活性的变化,如图3所示。据观察,对于Pd1@Pt2/C, 在第一次重复中,活性略有增加,可能是由于在过滤过程中彻底清洗了催化剂中的杂质Cl-离子。值得注意的是,随着重复次数的增加,Pt/C的转化率呈下降趋势,在10次迭代后下降了22%。此外较高氧化态(Pt2+和Pt4+)的Pt比例显著增加,从54.2%增加到66.6%。此外,在所用Pt/C的S核能级光谱中,在169eV出现了一个新的2p峰,归因于金属硫酸盐。这就解释了Pt在较高氧化态中所占比例增加的原因。因此,Pt催化剂性能的下降可能是由于氧化Pt含量的增加。相比之下,无论循环数如何,Pd1@Pt2/C的转化率曲线几乎保持不变,这表明Pd1@Pt2/C在整个重复使用过程中具有持续和稳定的催化活性。
2. 催化剂理化性质

图4 D-间距Pd1@Pt2
图4 显示了Pd1@Pt2/C的D间距,其中0.224nm和0.196nm分别对应于(111)和(200)平面,与XRD数据一致。由于Pt和Pd之间晶格参数的相似性,区分Pd和Pt元素具有一定难度
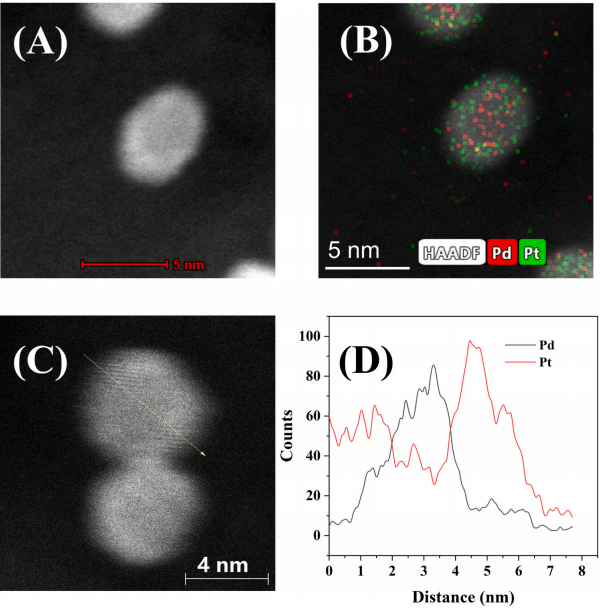
图5 (A)Pd1@Pt2/C的 STEM映射图像(B)Pd和Pt的重叠能量色散光谱;(C)和(D)Pd1@Pt2纳米颗粒的线扫描图像
利用高角度环形暗场(HAADF)和能量色散X射线光谱(EDS)测绘和线扫描技术,研究了Pd和Pt在Pd1@Pt2/C中的成分分布,如图5所示。在暗场图像中,观察到类似椭圆形的纳米粒子,显示出核和壳元素之间明显的对比差异。通过表面扫描和线扫描,可以看出纳米粒子包裹着Pd核的Pt壳,Pt壳厚度估计约为2.5nm。在制备过程中使用湿化学逐步沉积法,再加上Pd和Pt具有相似的面心立方(fcc)晶体结构和几乎相同的晶格常数,促进了Pt原子在Pd核上的外延生长,从而形成核壳结构的颗粒。

图6 催化剂的XRD图谱
图6描述了所制备的催化剂的XRD模式。对于所有催化剂,石墨载体的(002)面的衍射峰在2θ=24.9◦处观察到。在2θ值39.80◦ 、46.28◦和67.53◦ 处的突出衍射峰分别归因于面心立方(fcc)Pt(111)、(200)和(220)晶面的特征峰。此外,2θ值40.14◦、46.694◦和68.17◦ 处的峰分别被指定为fcc Pd(111)、(200)和(220)晶体平面的特征峰。尽管Pd和Pt之间的晶格参数略有不匹配(0.7%),但可以看出Pdx@Pty/C与Pt非常相似,间接肯定了Pt包裹在Pd周围的核壳结构。此外Pd1@Pt2/C催化剂的(111)峰位置相对于Pt/C催化剂表现出0.05◦的正移。这种位移比Pd1@Pt1/C催化剂大,表明Pt-Pt键的收缩归因于Pt壳和Pd核的晶格结构之间的不匹配。应变诱导的Pt 带中心下移对于提高Pt的催化活性至关重要。相反,Pd2@Pt1/C催化剂的(111)峰位置相对于Pt/C催化剂的负位移为0.07。
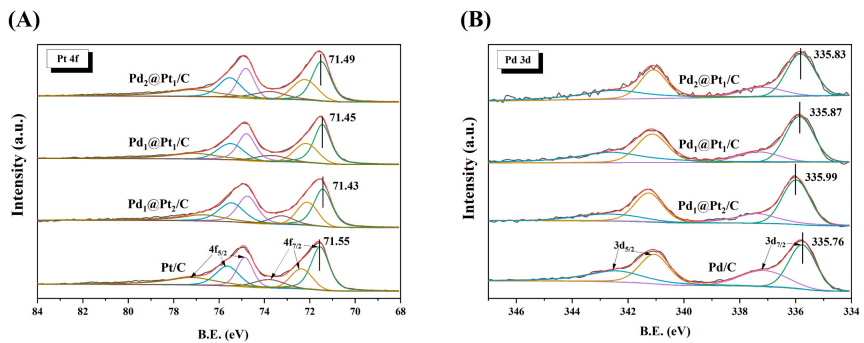
图7 催化剂的(A)Pt 4f和(B)Pd 3d的XPS光谱
为揭示表面电子变化对活性的影响,对催化剂进行了成分分析。催化剂Pt 4f和Pd 3d区域的XPS光谱如图7所示。Pt 4f在高结合能和低结合能区域的峰值分别对应于Pt 4f5/2和4f7/2态。每个主峰都可以解卷积为三个子峰,分别对应Pt(0)(71.43–71.55,74.75–74.86 eV)、Pt(II)(72.11–72.37,75.45–75.63 eV)和Pt(IV)(73.24–73.8,76.7–77.2 eV)。Pt(II)和Pt(IV)的存在导致Pt(0)的分离自旋轨道分量(3.31-3.34eV)低于Pt金属的特征值(Δ=3.35eV)。在Pd 3d区域的高结合能和低结合能处观察到的峰分别指示Pd 3d3/2和3d5/2区域。每个主峰可以解卷积为两个子峰,分别表示Pd(0)(335.76–335.99,341.06–341.26 eV)和Pd(II)(337.13–337.39,342.43–342.67 eV)。与Pd金属的特征值(Δ=5.26 eV)相比,Pd(II)的存在略微提高了Pd(0)的分离自旋轨道分量(5.27–5.3 eV)。与Pd/C相比,Pdx@Pty/C的Pd3d5/2峰值在向更高的结合能偏移。结合能随着Pd与Pt摩尔比的增加而增加。Pdx@Pty/C催化剂中Pd和Pt的结合能在表明核中的Pd和表面上的Pt之间存在强烈的电子相互作用,主要由Pd和Pt的电负性决定。由于Pt的电负性(2.28)高于Pd(2.2),因此Pd与Pt之间有明显的电子转移倾向,导致Pt 4f峰的负移和Pd 3d峰的正移,与之前的文献一致。电荷转移随后导致Pt中更高d带的填充,从而导致费米能(EF)的增加和Pt εd的向下偏移。d带中心的向下移动削弱了甲酸氧化中CO*与Pt的结合,从而增强了催化剂的抗中毒性。
3.催化剂的电化学性能
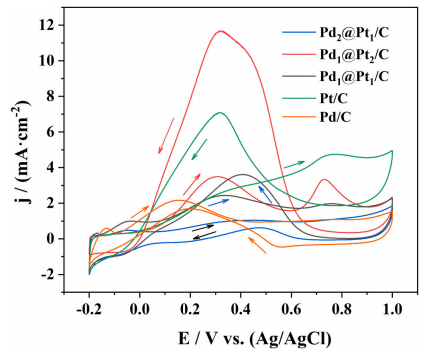
图8 扫描速率为50 mV⋅s−1时,各种催化剂在0.5 M甲酸和0.5 M H2SO4溶液中的CV曲线
利用循环伏安法(CV)研究了催化剂对甲酸(FA)氧化的催化途径。图8描绘了以下物质的循环伏安图:Pdx@Pty/C、 Pt/C和Pd/C在0.5M甲酸和0.5M H2SO4中的溶液。观察到,在正向扫描过程中,Pt/C在0.77 V处显示出明显的氧化峰,在0.4 V左右出现肩峰。0.77 V的电势落在与甲酸氧化间接途径相关的电化学范围内。甲酸在Pt/C上的氧化主要通过CO途径进行,如下所示:
M + HCOOH→M-CO + H2O→M + CO2 + 2H+ + 2e− (1)
对于Pd/C的CV曲线,在0.16 V处观察到一个主要的氧化峰,在-0.13 V附近有一个明显的氢吸附峰,这表明甲酸氧化主要遵循Pd/C上的直接途径,如下所示:
M + HCOOH→X→M + CO2 + 2H+ + 2e− (2)
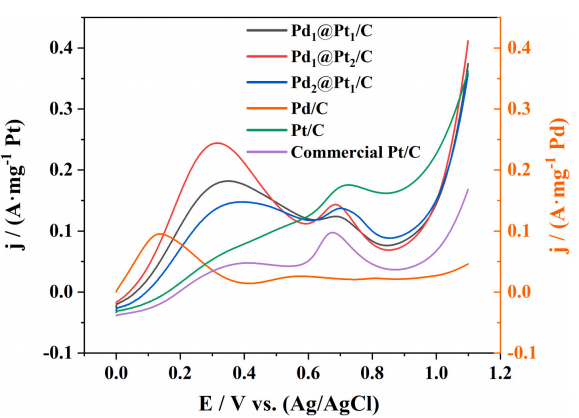
图9 不同催化剂在0.5 M HCOOH和0.5 M H2SO4溶液中的LSV曲线,扫描速率为10 mV⋅s−1
在Pd1@Pt2/C和Pd1@Pt1/C的CV曲线中观察到两个强度相当大的不同氧化峰。值得注意的是,在0.31-0.34 V附近观察到的峰归因于甲酸氧化的直接途径(即脱氢途径),而位于0.73-0.76 V附近的峰对应于甲酸氧化中的间接途径(即脱水途径)。与Pd/C类似,Pd2@Pt1/C的氧化电位较低,初级氧化峰出现在0.45V,表明甲酸氧化主要通过“直接途径”发生。图9显示了在0.5 M甲酸和0.5 M H2SO4中甲酸(FA)氧化的Pdx@Pty/C、Pt/C、Pd/C和商用Pt/C的线性扫描伏安(LSV)图。Pd1@Pt2/C在Pt负载下表现出最高的质量活性(MA),,并且与Pt/C相比,在较低电位下对甲酸氧化表现出明显的催化电流。Pd1@Pt2/C(0.244 A⋅mg−1 Pt)的最大质量活性是商用Pt/C(0.097 A⋅mg-1 Pt)的2.5倍,突显了Pd掺入Pt对甲酸氧化的促进作用。虽然高催化活性通常通过高质量活性来评估,但Pd/C是一个例外,其质量活性与商用Pt相似,但在实际催化中表现不佳。通常,去除催化剂表面吸附的CO*物种需要在相对较高的电势下进行操作。然而,对于Pd/C,甲酸的催化氧化主要通过直接途径进行,导致相对较少的CO*物种难以通过循环伏安法(CV)曲线直接检测。因此,CO*的积累和去除分析需要电位控制。
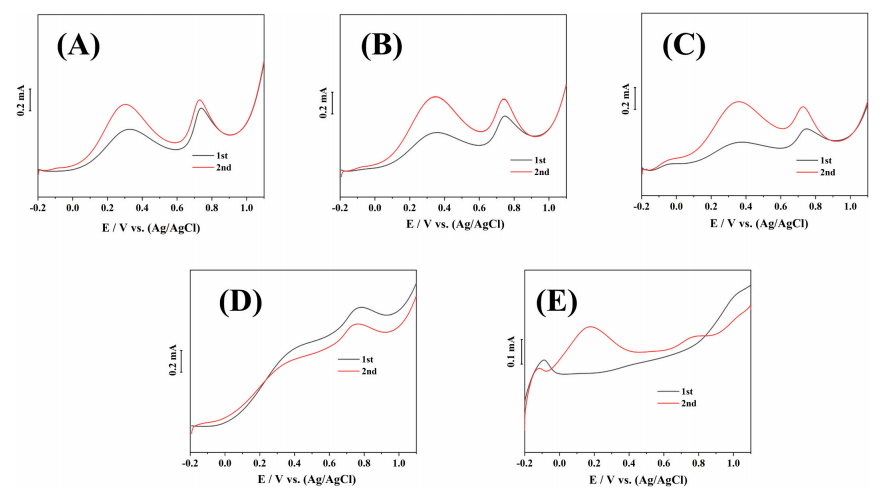
图10 (A)甲酸氧化性能恢复的比较评价Pd1@Pt2/C、 (B)Pd1@Pt1/C、 (C)Pd2@Pt1/C、 (D)在CO*积聚和去除后的Pt/C和(D)Pd/C催化剂
如图10所示,将上限电位设置为0.6 V,持续5个周期,以积累表面中毒物种。随后,将上限电位调节至1.1V,持续2个循环,以分析催化性能的恢复情况。在上限电位设置为1.1V的第一次正向扫描期间,Pdx@Pty/C和Pt/C表现出明显的甲酸氧化电流。相比之下,除了0 V以下的氢吸附峰外,Pd/C显示的甲酸氧化电流可以忽略不计,表明虽然甲酸氧化主要通过Pd/C上的直接途径进行,但CO*对Pd/C的催化活性具有明显的抑制作用。在第二次正向扫描中,表明Pdx@Pty/C和Pd/C在电流密度方面表现出明显的恢复,而Pt/C则经历了一定程度的下降,表明Pdx@Pty/与Pt/C相比, Pd/C具有较低的CO氧化电位。吸附在表面上的有毒物质在较低电位下产生,在较高电位下被去除。相反,Pt/C上的甲酸氧化主要通过间接途径进行,导致产生更高丰度的CO*物种,而1.1 V电压上限无法完全消除CO*物种。
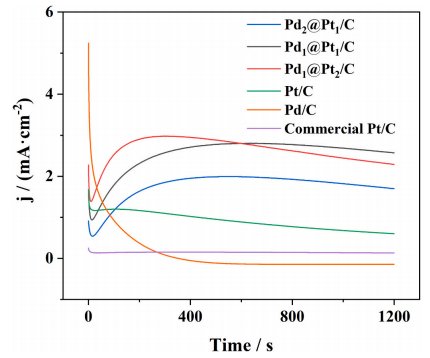
图11 不同催化剂在0.5 M甲酸和0.5 M硫酸溶液中在0.25 V下的CA曲线
为评估催化剂在甲酸氧化反应(FAOR)中的耐久性,对Pdx@Pty/C、 Pt/C和Pd/C进行了计时电流法(CA)测量,记录1200秒的结果并在图11中进行比较。对于Pd/C催化剂,它表现出最高的初始电流,但迅速下降,最终稳态电流接近0A,进一步证实了中毒物种对Pd/C催化的FAOR的抑制作用以及Pd/C催化剂在V3.5+电解液催化生产过程中的失活。在1200秒内,Pt/C催化剂的电流密度从最初的1.68 mA⋅cm−2降低到0.60 mA⋅cm-2,而商业Pt/C催化剂则从0.25 mA⋅cm-2下降到0.13 mA⋅了cm−2中。值得注意的是,对于Pdx@Pty/C催化剂,在初始快速下降后,氧化电流密度明显增加,表明Pdx@Pty/C优异的抗CO中毒性。具体而言Pd1@Pt2/C表现出卓越的稳定性,具有最高的初始电流(2.28mA⋅cm−2),而1200秒后的氧化电流保持在2.29mA⋅cm-2。在文献中,有报道称,在Pd表面上,通过“直接途径”机制产生的-COOH物种易于与氢原子反应(用于HCOOH分解),形成有毒的CO,从而抑制HCOOH的氧化。
4.VRFB性能
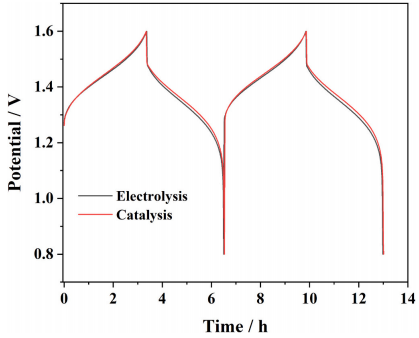
图12 使用通过催化和电解在80 mA⋅cm−2下制备的V3.5+电解液的电池的充放电曲(0.8–1.6 V)
将V3.5+电解液引入单个VRFB电池进行充放电测试,以评估电解质性能,将通过催化还原制备的电解质与Pd1@Pt2/C进行比较。结果如图12所示。用Pd1@Pt2/C制备的电解液的CE和EE(CE:94%,EE:86%)与电解制备的电解液的几乎相同。
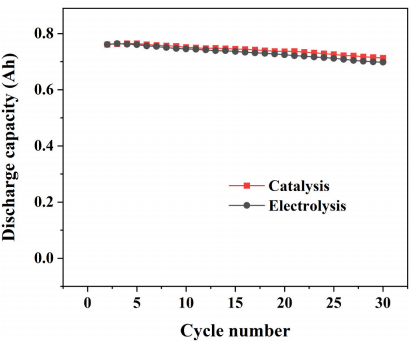
图13 使用通过催化和电解制备的V3.5+电解质在120 mA⋅cm−2下的电池循环放电容量曲线
此外,为更好地说明电池中电解液的性能,图13显示了循环时的放电容量曲线。催化法制备的电解液和电解制备的电解液表现出几乎相同的容量逐渐下降,主要受钒离子交叉的影响,验证了通过催化还原合成的电解液与传统电解液的质量相当。
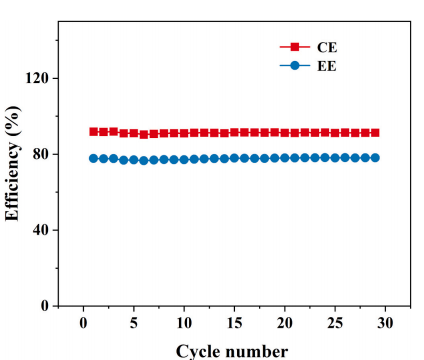
图14 利用通过催化产生的V3.5+电解液的电池的库仑和能量效率
此外,通过催化还原制备的V3.5+电解液在0.80-1.60 V范围内以120 mA cm−2的电流在30个循环内表现出稳定的CE和EE(图14),表明电解液的稳定性。
结论展望
浙江大学高翔院士&宋浩教授团队开发了Pd@Pt/C催化剂用于催化还原制备VRFBs的V3.5+电解液。就催化性能而言,Pd1@Pt2/C对用作还原V4+的还原剂的甲酸表现出优异的氧化活性。Pd1@Pt2/C催化剂的甲酸氧化活性比商业Pt/C的高出2.4倍。得益于Pd和Pt之间的协同作用,甲酸氧化过程中CO中间体的吸附得到缓解,使催化剂对CO中毒的抵抗力增强。此外,Pd的掺入改变了甲酸在Pt/C上的间接氧化途径,从而增强了其催化效果。Pt壳可以保护Pd在V3.5+电解液的催化生产中免于失活。在重复生产中,Pd1@Pt2/C 保持了效率而没有退化,从而产生了性能优异的电解液。因此,该催化剂有望高效稳定地生产用于VRFB的V3.5+电解液。
文献信息
Zhesheng Hua , Li Wang , Hao Song , Xiao Zhang , Chenghang Zheng , Shaojun Liu , Yang Yang ,Xiang Gao,A promising catalyst for efficient and stable production of high-performance V3.5+ electrolyte in vanadium redox flow batteries.2024, Journal of Energy Storage
https://doi.org/10.1016/j.est.2024.113164